The complexity of enzyme catalysis, consisting of ligand recognition and exquisite discrimination followed by rapid catalytic turnover, has traditionally been difficult to study. Recent developments in molecular modelling of macromolecules and powerful molecular biological methodologies now offer new approaches for improving our understanding of enzyme structure-function relationships. Our research program combines these methodologies to explore the fundamentals of enzyme catalysis. The benefits of better understanding enzyme catalysis are numerous: we are gaining a better understanding of enzyme-based drug resistances and are modifying enzymes for synthetic applications. Currently, there is a pressing for development of complementary strategies to provide greater detail about the nature of enzyme-ligand interactions for applications including drug design and biocatalysis. To improve our capacity to modify enzymes for synthetic purposes, we are gaining additional insight into the plasticity of enzyme active sites by distinguishing the elements that are indispensable for function from those that can be altered. We develop and apply methodologies that enhance the speed, power and information-content of the various steps implicated in enzyme engineering.
Transglutaminases (TGase) catalyse the formation of crosslinks between proteins or peptides. In humans, the TGase-mediated cross-linking of proteins is implicated in blood clotting, endocytosis, apoptosis and can also lead to physiological disorders such as cataract formation and Celiac disease. It is thus a target for drug design. In a long-standing collaboration with Professor Jeffrey Keillor (Ottawa U.), we generated a model for the binding of small acyl-donor peptide substrates to mammalian tissue transglutaminase toward the development of model substrates and inhibitors (Chica et al. (2004) Prot. Sci. 13(4):979-991), and explored the use of non-native substrates (Keillor et al.(2008) Canadian J. Chemistry,86, 271-276. Making use of new assays such as this fluorometric assay for transamidation activity (Gnaccarini et al. (2009) . Bioorg Med Chem., 17 (17): 6354-9) we discovered potent cinnamoyl inhibitors of mammalian TGase (Pardin et al.(2009) Biochemistry 48 (15): 3346-53; Pardin et al. (2008) J. Organic Chemistry, 73, 5766-5775.
In a bid to develop its use as a biocatalyst, we also demonstrated site-specific protein propargylation using mammalian tissue transglutaminase (Gnaccarini et al.(2012) Org. Biomol. Chem.DOI: 10.1039/C2OB25752A). We have more recently turned to microbial transglutaminases (MTG) as a green alternative to amide synthesis. MTG is widely used in the food and textile industries. MTG is tolerant to a broad range of reaction conditions and is therefore suitable for further development as a biocatalyst. We investigated a wide range of non-natural, small-molecule substrates, which were chosen to explore enzyme reactivity and to yield synthetically relevant products. Our results indicate that a variety of amines function as an acceptor substrate, but stricter requirements apply for the donor substrate. Importantly, we demonstrated that MTG can be used to add an alkyne or an azide onto a peptide chain, for ulterior 'Click' reaction (Gundersen et al., Appl Microbiol Biotechnol DOI 10.1007/s00253-013-4886-x).
Team members:
__________________________________________________________________________________________________
Biomarkers are specific molecules that correlate with the progression of diseases or with the susceptibility of a disease to a given treatment. They are useful indicators for monitoring the state of progression or the treatment of a particular disease. We are developing protocols for monitoring different cancer-related biomarkers using surface plasmon resonance (SPR). The attachment of a particular protein of interest on gold surface allows for specific detection of protein-protein interaction or binding of a target molecule of interest using SPR. For example the attachment of an antibody on a gold surface allows for the detection of its specific antigen in clinical samples or the attachment of a specific antigen on the surface may allow for monitoring the level of antibody, a useful indicator of the course of treatment of many diseases.
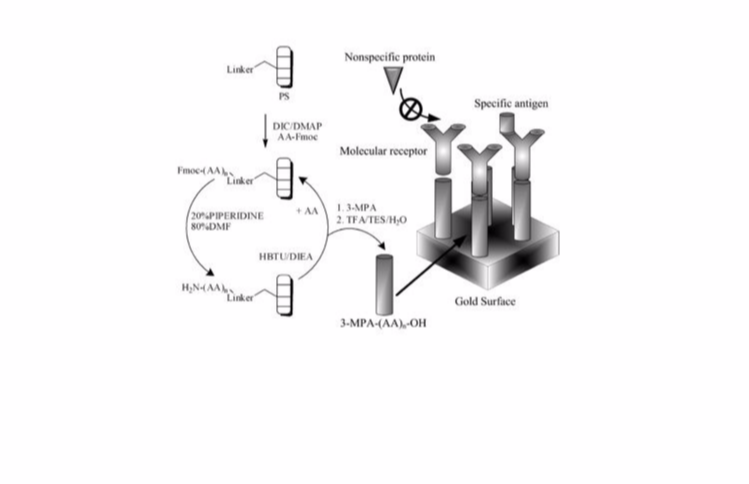
We are developing protocols adaptable to a new portable SPR device designed by Professor Jean-François Masson. We developed and patented different types of surface monolayers preventing non-specific adsorption that allow for SPR detection of specific molecules using crude serum (Ratel et al., Anal Chem 2013, 85, 5770-5777; Aubé et al., Langmuir 2013, 29, 10141-10148; Bolduc et al., Analyst 2011, 136, 3142-3148; Bolduc et al., Anal Chem 2010, 82, 3699-3706; Bolduc et al., Anal Chem 2009, 81, 6779-6788). We employ this approach for monitoring methotrexate in clinical samples of cancer patients (Zhao et al., Analyst 2012, 137, 4742-4750). We are now developing different new SPR assays for monitoring cancer-related biomarkers and bacterial infections. We are working on improving the sensitivity of detection by optimizing both the physical and the biological aspects of the SPR system. Our work includes protein engineering and surface chemistry to work out new SPR protocols for improving the accessibility of diagnosis by monitoring the progression, the course of treatment or the screening of different diseases.
Team members:
__________________________________________________________________________________________________
The continuously increasing use of trimethoprim as a common antibiotic for medical use and its preventive application in terrestrial and aquatic animal farming has increased its prevalence in the environment. This has been accompanied by increased drug resistance, generally in the form of alterations in the drug target, dihydrofolate reductase (DHFR). The most highly resistant variants of DHFR are known as type II DHFRs, among which R67 DHFR is the most broadly studied variant. It is imperative to discover inhibitors of this new source of antibiotic resistance, to maintain the efficacy of trimethoprim in treatment of diseases including pneumonia, gonorrhea and adventitious infections in AIDS patients.
We have reported the first specific inhibitors of this emerging drug target, obtained by fragment-based design (Bastien et al. (2012) J. Med. Chem. 55, 3182-3192.). We developed a functional screening platform for the identification of selective inhibitors of the bacterial antibiotic-resistance enzyme, R67 DHFR. The platform is based on semiautomated determination of enzymatic activity in the presence of a variety of fragments, or simple compounds similar to the native ligands of the enzyme, to assess inhibition. We observed that a variety of small aromatic compounds offer millimolar range inhibition of R67 DHFR, which is consistent with the proposed “primitive” nature of its relatively promiscuous binding site (Schmitzer et al. Protein Eng. Des. Sel., 11, 17, 809-819 (2004)). By those means, small aromatic molecules of 150−250 g/mol (fragments) inhibiting R67 DHFR selectively in the low millimolar range were identified. Weakly inhibiting molecules provided the basis for testing compounds of greater complexity, which provided an increase in affinity from millimolar to micromolar. More complex, symmetrical bis-benzimidazoles and a bis-carboxyphenylprocured selective inhibition of the target in the low micromolar range (Ki = 2−4 μM). Molecular docking using our high-resolution crystal structure (Yachnin et al. (2011) Acta Crystallogr. F 67, 1316-1322) offered insights into the binding mode of these inhibitors. Importantly, we screened the human DHFR in parallel with R67 DHFR to identify compounds with the best prospects for selective inhibition. Thus, we identified a new class of selective, symmetrical, and competitive inhibitors of R67 DHFR.
We are currently working on the next generation of R67 DHFR inhibitors.
Team members:
__________________________________________________________________________________________________
Beta-lactamase production has become a world-wide contributor of bacterial resistance to widely-used clinical antibiotics such as penicillins and cephalosporins. Class A beta-lactamases have become extensively studied model enzymes. The high rate of occurrence of mutated enzymes capable of hydrolysing higher generation cephalosporins has stimulated research on the mechanisms of beta-lactamase adaptation to these new substrates, to understand the molecular basis of this evolutionary chain of events. By performing directed evolution where concurrent mutations were focussed within the active-site cavity, we observed a high tolerance to simultaneous active-site mutations in TEM-1 beta-lactamase, as well as distinct mutational paths which provide more generalized substrate recognition (P.-Y. De Wals et al. (2009) Protein Science, 18 (1): 147-160).
By way of mutagenesis, enzyme kinetics, protein NMR dynamics and molecular simulations, we showed that the conserved residue Tyr105, which delineates one of the edges of the active site cavity, serves as a 'gate-keeping' residue, conferring selectivity for the incoming substrates. The adopted conformation of residue 105 prevents detrimental steric interactions with the substrate in the active site cavity and provides a rationalization for the strong aromatic bias found in nature at this position among Class-A beta-lactamases. (N. Doucet, P.-Y. De Wals and J. N. Pelletier (2004) J. Biol. Chem. 279 (44): 46295-46303). We performed simulated annealing of tyrosine 105 in TEM-1 ß-lactamase. It suggested the existence of alternate conformations (N. Doucet and J. N. Pelletier (2007) Proteins, 69, 340-348). These were further investigated using protein NMR spectroscopy. The number and magnitude of short and long range effects on (1)H-(15)N chemical shifts were correlated with the catalytic efficiencies of Tyr105X mutants. In addition, (15)N relaxation experiments revealed that several active-site residues display altered motions on both picosecond-nanosecond and microsecond-millisecond time scales despite many being far away from the site of mutation. The altered motions may account for the decreased catalytic efficiency, suggesting that short and long range residue motions could play an important catalytic role in TEM-1 beta-lactamase (N. Doucet et al. (2007) J. Biol. Chem., 282, 21448-21459).

To better understand , we investigated laboratory-evolved 'chimeric' beta-lactamases, that are constituted of alternating blocks from TEM-1 and fromthe contribution of dynamics to catalysis in beta-lactamases PSE-4 beta-lactamases (Voigt et al., Nat. Struct. Biol. 2002 9:553). We characterized a series of functional TEM-1/PSE-4 chimeras possessing between 17 and 92 substitutions relative to TEM-1 β-lactamase. Circular dichroism and thermal scanning fluorimetry revealed that the chimeras were generally well folded. Despite harbouring important sequence variation relative to either of the two 'parental' β-lactamases, the chimeric β-lactamases displayed substrate recognition spectra and reactivity similar to their most closely-related parent (Clouthier et al. (2012) PLoS One. 7(12):e52283). NMR backbone resonance assignments confirmed the native-like fold of the chimera with 17 substitutions, consistent with its native-like activity (S. Morin et al. (2010) Biomol NMR Assign., 4 (2):127-30). To gain further insight into the changes induced by chimerization, that chimera was investigated by NMR spin relaxation. While high order was conserved on the ps-ns timescale, a hallmark of class A β-lactamases, evidence of additional slow motions on the µs-ms timescale was extracted from model-free calculations. This is consistent with the greater number of resonances that could not be assigned in this chimera relative to the parental β-lactamases, and is consistent with this well-folded and functional chimeric β-lactamase displaying increased slow time-scale motions (Clouthier et al. (2012) PLoS One. 7(12):e52283).
__________________________________________________________________________________________________
ADRESSE / ADDRESS
Université de Montréal
Département de Chimie
Campus MIL - Local B-6227
1375 Ave.Thérèse-Lavoie-Roux
Montreal, Qc, Canada, H2V 0B3
CONTACT
Phone: +1 (514) 343-2124
Fax: +1 (514) 343-7586
@ : joelle.pelletier@umontreal.ca